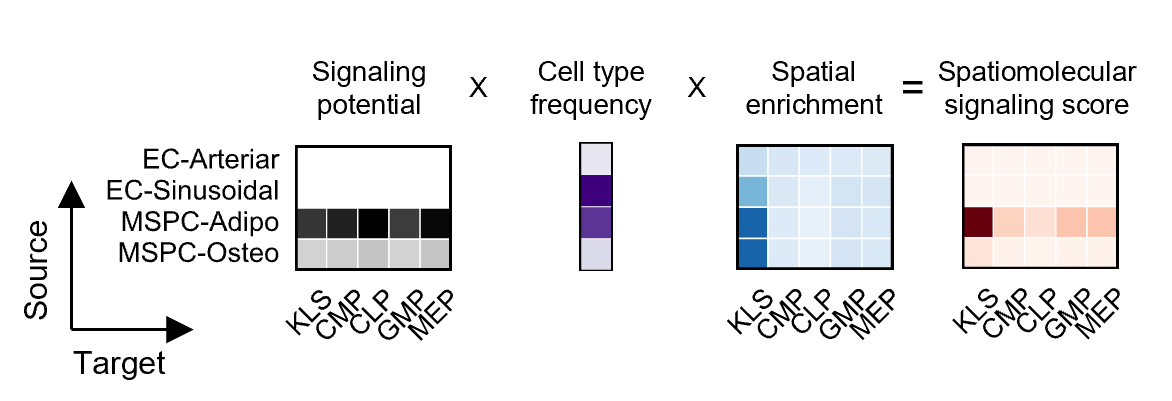Intercellular Signaling Networks
Intercellular interactions in HISE are all based on scRNA-seq data. This data is inherently void of any spatial information. Further, these data do not accurately reflect the proportional make up of the bone marrow environment. Here, we attempt to elucidate intercellular signaling patterns taking into account the proportional make up of the bone marrow as well as spatial organization.
How networks were constructed
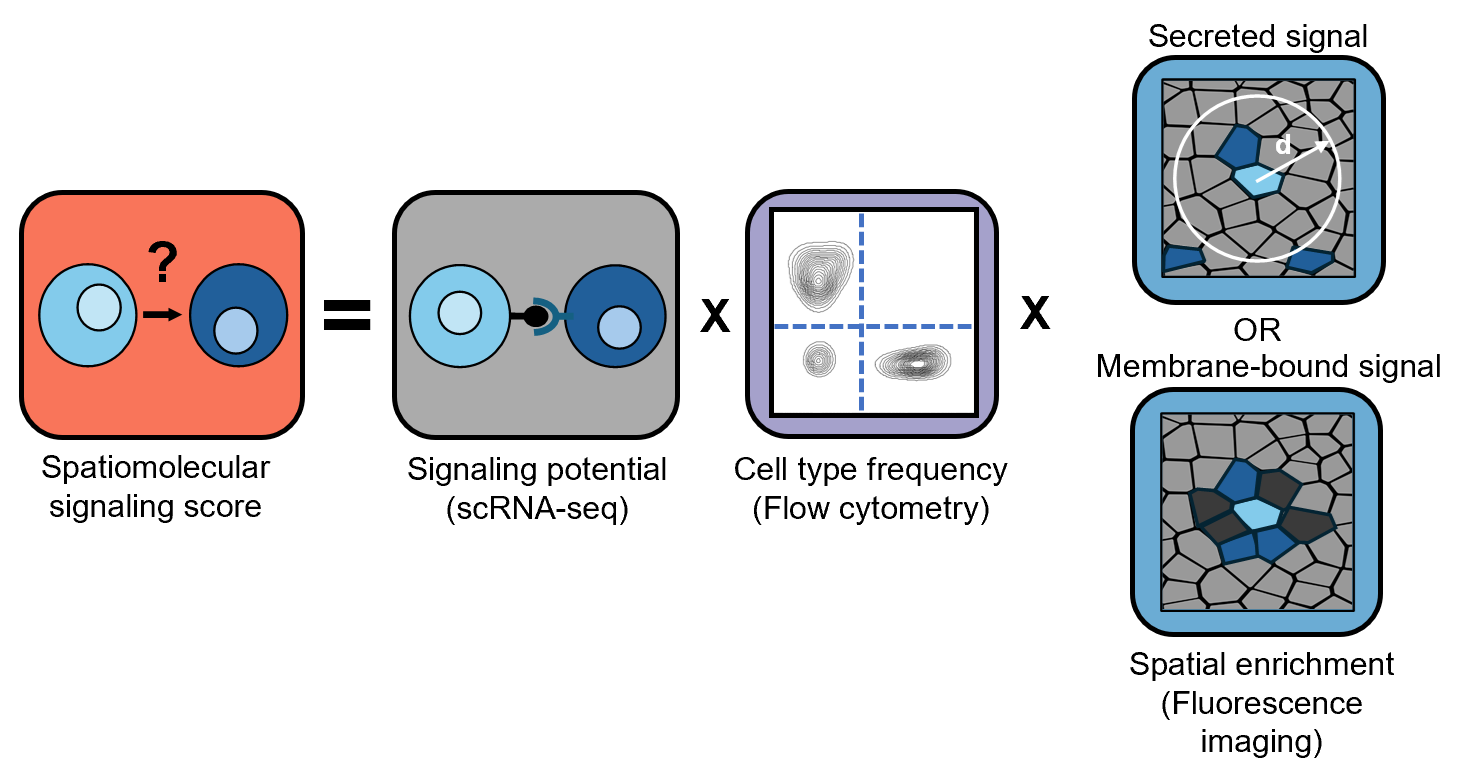
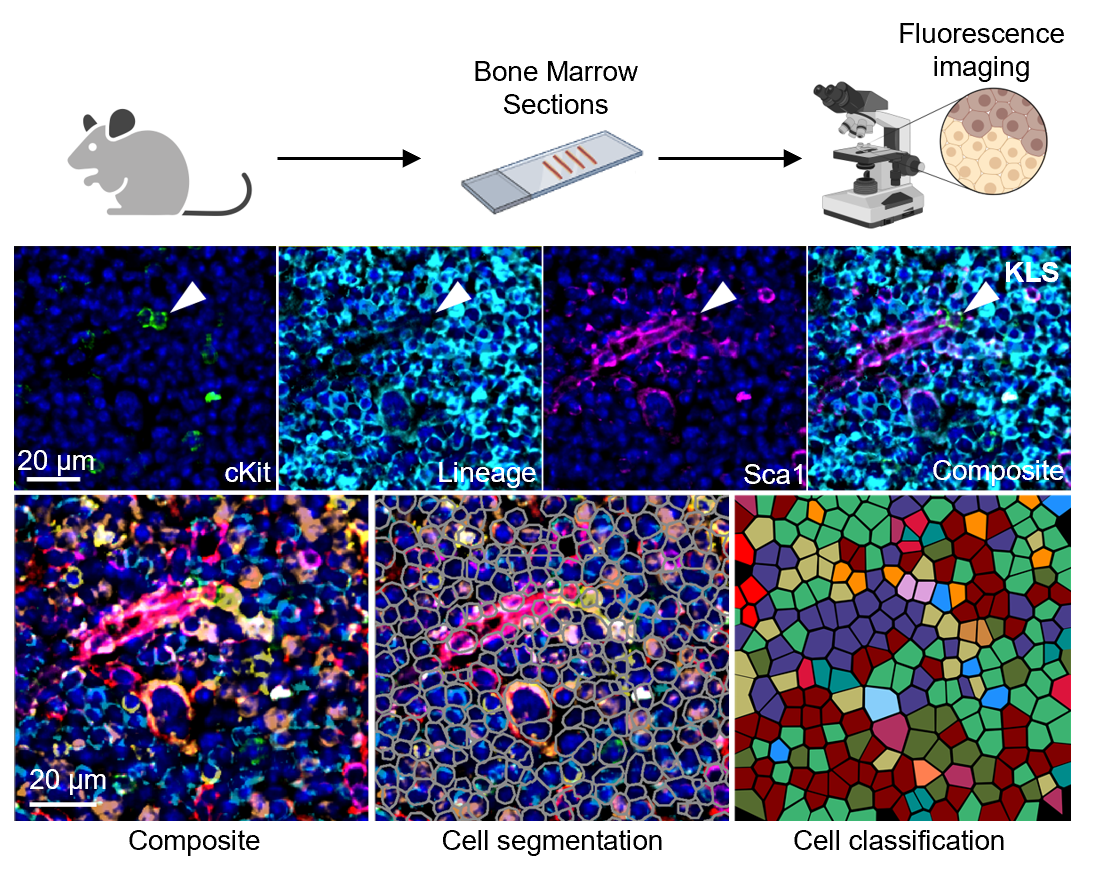
Signaling networks were constructed using data from three sources: scRNA-seq, FACS, and Phenocycler. Using CellChat on scRNA-seq data, we calculate interaction intesnity values between HSPCs and other cell types. These interaction intesnity values are then adjusted by FACS, data that reveals cell type fraction in the bone marrow. Finally, these values are adjusted by spatial data from the Phenocycler, specifically, which cells are found within a 20 μm distance from an HSPC (for secreted signaling pathways) or cells that are found in direct contact with an HSPC (for membrane-bound signals) at rates significantly higher than random chance.
Data for all cell types identified by scRNA-seq is not always obtainable by the other measurement strategies and therefore, at each step, the model is restricted by the cell types that are identifiable with the given technologies. For example, with our current set-up, HSCs and MPPs are not identifiable in Phenocycler data, but KLS (cKit+Lin-Sca+) cells are. In the network diagrams,
Phenocycler Imaging
Sample Calculation: MK Pathway